


Predicting protein conformational changes remains a crucial challenge in computational biology and artificial intelligence. Breakthroughs achieved by deep learning, such as AlphaFold2, have moved the goalpost for predicting static structures but do not address the dynamic conformational change most proteins undertake to exercise their biological roles. These transitions are critical to understand a wide range of biological processes from enzyme activity to signal transduction. However, the lack of structural data for intermediate states hampers the prediction of these transitions. Furthermore, the existing models suffer from high free energy barriers of transition states, making accurate predictions even more challenging. Catalytic for the advancement of a range of fields, including drug design, synthetic biology, and disease research, it will be theÂ
The existing models to describe protein conformational transitions include elastic network-based normal mode analysis, as well as hybrid models that combine elastic networks with molecular dynamics simulations. These methods are appropriate for fairly simple conformational movements but do not have the resolution to account for the complex and vast changes found in larger proteins. More recently, deep learning approaches, such as auto-encoders, Boltzmann generators, and diffusion models, have been developed, mapping protein structures onto low-dimensional latent spaces. However, these models depend on a linear pathway between two states, which does not apply in complex, nonlinear transitions, such as fold-switching. More importantly, the high data demands and low efficiency of the data, in addition to a computational cost that precludes real-time scalable applications, make these approaches themselves unsatisfactory.
The authors detail a novel deep learning strategy by making use of high-throughput biophysical sampling to circumvent the protein conformational transition-related data paucity. Molecular dynamics simulations were combined with enhanced sampling methods to produce a library of 2,635 proteins with two experimentally determined states. This dataset uses an overall deep learning model that is called PATHpre which predicts structural pathways that result in conformational transitions with high accuracy. Centrally, the innovation to the HESpre module in PATHpre pertains to the predictive performance of the high-energy state along the transition pathway. The proposed model makes no linear latent space assumptions that could be a subject of criticism. It presents immense generalizability toward proteins of diverse conformations. That would mean a huge contribution it addresses the dynamic behavior modeling within complex systems, applying scalability and data efficiency at an approach level.
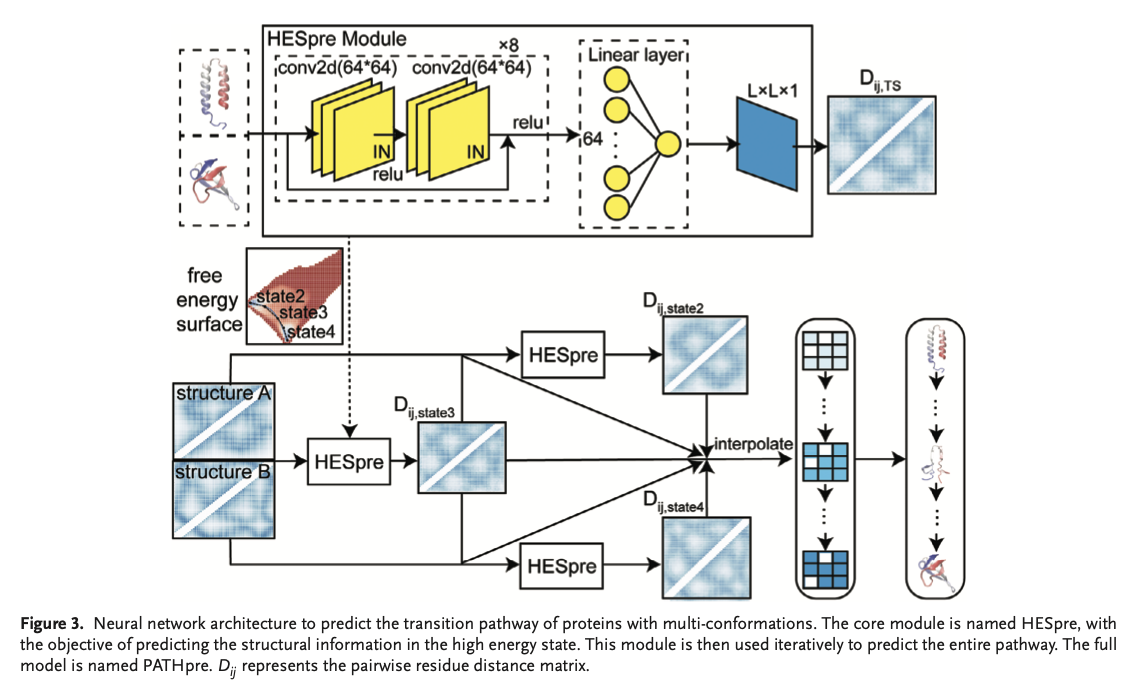
In a PATHpre approach, distance matrices in a two conformation states system are applied through convolution neural network prediction to acquire a high-energy state between such conformational states; that is where HESpre dwells on: only special or unique contacts that it ascertains are taking place in high energy of the residue pair across each pathway based on the pairwise distance matrix quantifies contact formation and rupture for pathway taken and the overall contact matrix established. It contains four classes of classified MS proteins that depict inter-domain and intra-domain movements, localized unfolding, and global fold changes in their conformational properties. Cross-validation on various proteins was performed for the model, which achieved strong Pearson correlations and low mean absolute errors at all steps; thus, it is very versatile across structural classes. The good performance generally establishes the general applicability of the model across proteins of different sequence lengths and complexities of structures.Â
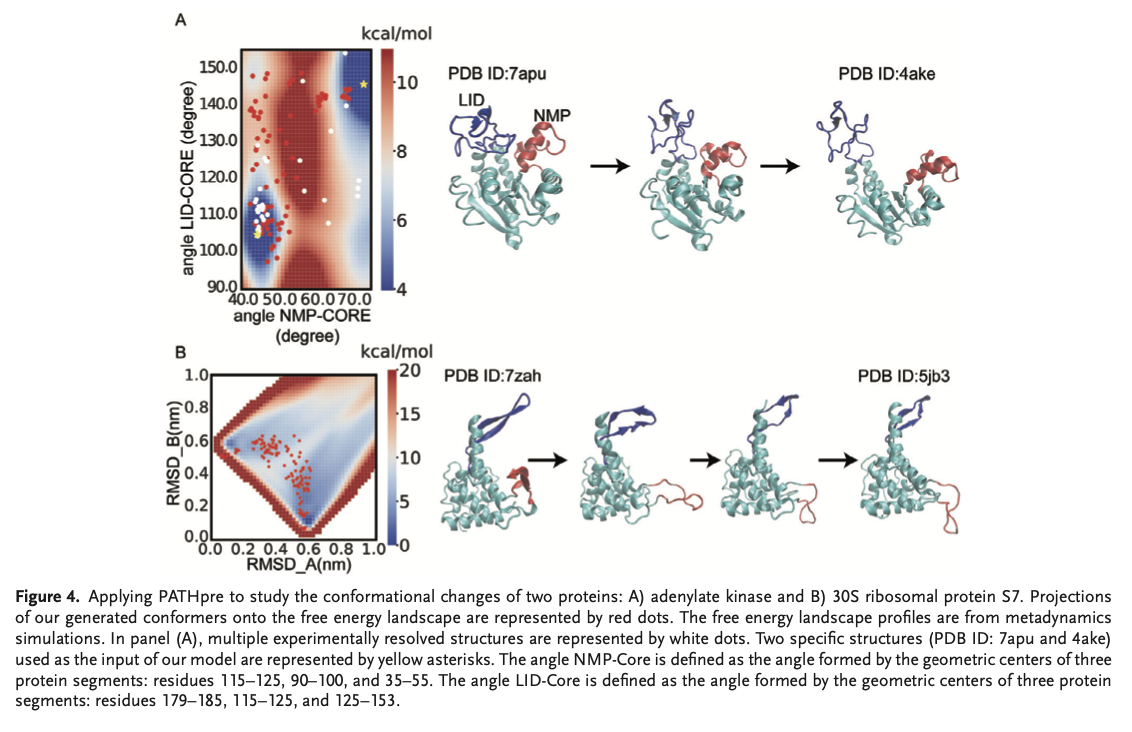
PATHpre is accurate in the very high predictions of protein transition pathways by displaying strong correlations with the experimental and simulated data that exist on a range of proteins. Evaluations also showed that PATHpre robustly captures simple to complex conformational changes and it is consistent to varying lengths of sequence as well as structural complexity. Importantly, it accurately predicted the transition pathways for individual proteins, such as adenylate kinase and the 30S ribosomal protein S7, by matching the experimental free energy landscapes and performed better than conventional hybrid approaches in challenging conditions. PATHpre predictions were aligned with the known structures, and its mapping of fine intermediate states in fold-switching proteins confirmed its wide applicability and reliability to capture the vast spectrum of protein conformational transitions.Â
This work marks significant progress in AI-driven protein modeling, providing a data-efficient and scalable approach toward predicting protein conformational transitions. The integration of large-scale biophysical sampling with deep learning in PATHpre addresses the most stringent challenge of limited data and captures nonlinear transitions across the diversity of proteins. This generalizable model will indeed form the basis for the greatly enhanced application of AI applications in computational biology, establishing thus a powerful tool for investigating dynamic protein behavior within a range of contexts- from drug discovery to synthetic biology.Â
Check out the Paper. All credit for this research goes to the researchers of this project. Also, don’t forget to follow us on Twitter and join our Telegram Channel and LinkedIn Group. If you like our work, you will love our newsletter.. Don’t Forget to join our 55k+ ML SubReddit.
[Sponsorship Opportunity with us] Promote Your Research/Product/Webinar with 1Million+ Monthly Readers and 500k+ Community Members
The post A Study on Protein Conformational Changes Using a Large-Scale Biophysical Sampling Augmented Deep Learning Strategy appeared first on MarkTechPost.
Source: Read MoreÂ

